Note from Andrew: This week’s piece fromRuxandra Teslo and Jack Scannell helped me understand the barriers to having more and better drugs. Ruxandra is a fellow at Renaissance Philanthropy where she studies how to improve clinical development. Jack is CEO of Etheros Pharmaceuticals Corp.
Before you hop into Ruxandra and Jack’s piece, I wanted to share an opportunity for current or recently graduated PhD students. IFP has partnered with some of the best economists of science and innovation to create a free online course called the Economics of Ideas, Science, and Innovation. Apply by January 9.
OK — let’s learn about clinical trials.
Introduction
Public debates about how to revive productivity in the biopharmaceutical industry tend to be dominated by two camps. Technological optimists usually argue that declining industry outputs relative to investment reflect gaps in biological knowledge, and that advances in basic science will eventually unlock a wave of new therapies. The second camp, which traces its intellectual lineage to libertarian economists, focuses on easing the burden of regulation. In their view, excessive FDA caution has slowed innovation. They propose solutions that largely target regulatory approval: either loosening evidentiary standards or narrowing the FDA’s mandate to focus solely on safety rather than on efficacy.
Both perspectives contain some truth. Yet by focusing on the two visible ends of the drug discovery pipeline, early discovery and final approval, both camps miss the crucial middle: clinical development, where scientific ideas are actually tested in people through clinical trials. This stage is extraordinarily expensive, operationally intricate, and crucially, generates the field’s most consequential evidence. We believe that systematic optimization of this middle stage offers significant untapped leverage and deserves far greater focus.
The need for such a shift becomes clearer when we consider the growing divergence between scientific potential and clinical results. Over the past few decades, biomedical science has advanced at a staggering pace. Genetics has moved from single-gene studies to sequencing the first human genome, a 13-year, $3-billion project completed in 2003, to today’s ability to read an entire genome in a day for a few hundred dollars. Protein science has followed a similar trajectory: from the earliest crystallographic structures in the 1950s, to large-scale structural biology pipelines, to AI tools like AlphaFold that can now predict protein structures computationally.
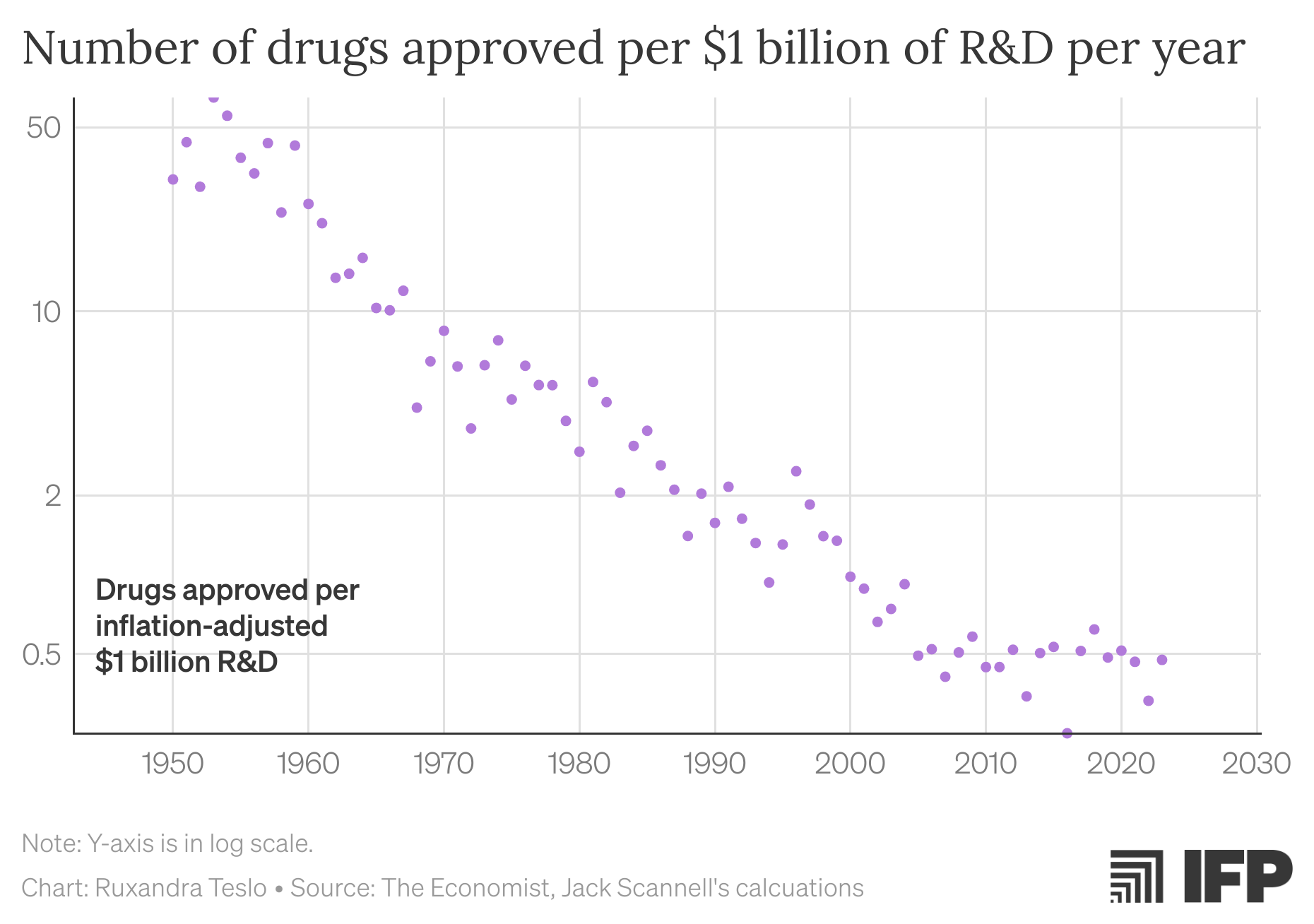
Yet, paradoxically, drug discovery has become increasingly inefficient. This trend, first identified in 2012 by one of the authors of this piece and termed Eroom’s Law (the inverse of Moore’s Law), describes how the inflation-adjusted cost of developing a new drug had doubled approximately every nine years since the 1960s. More recent data suggests that drugs per billion dollars spent has plateaued, while financial returns have continued to decline as new drugs tend to be approved for use in smaller groups of patients.
Figure 1. Pharmaceutical R&D productivity has steadily decreased since 1960, despite advances in basic science. In the last decade, the deceleration in pharmaceutical productivity seems to have ameliorated, due to a combination of factors including an increase in predictive validity (through e.g., genetics), but also due a larger share of efforts being directed at oncology and rare diseases, where the burden for approval is often lower due to the high unmet need.
Declining returns on R&D are no longer the sole concern; intensifying competition from China has amplified the urgency for change. As a Time magazine headline in May 2025 warned, “The US can’t afford to lose the biotech race with China.” Like many such commentaries, it calls for reform and renewed attention on the question of what the US can do to compete in the current international landscape.
But where along the drug development pathway do we have the greatest opportunity to steer things differently?
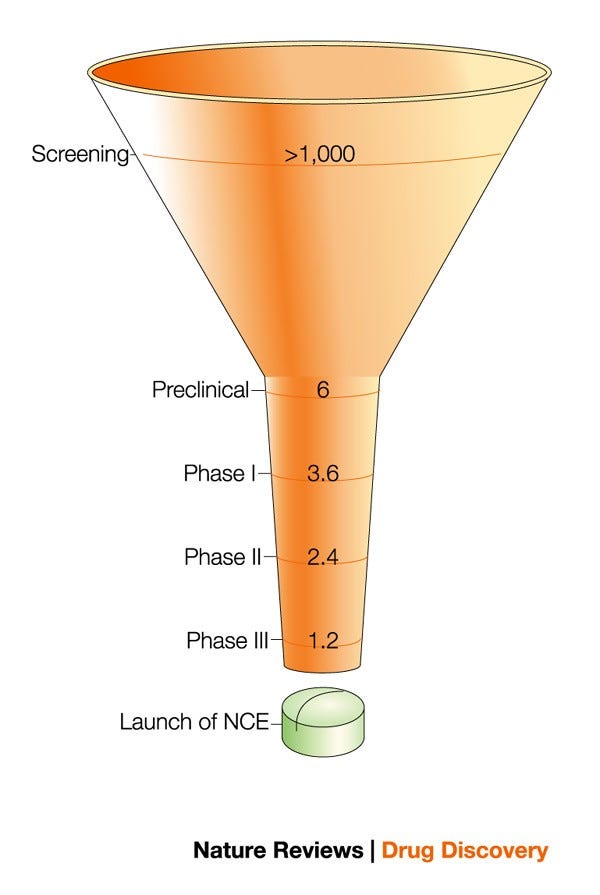
Drug discovery can be thought of as a funnel: broad at the start and progressively narrowing toward approval. Feeding into the funnel at one end stands basic science, which generates countless hypotheses. Only a fraction of these will survive preclinical validation and enter clinical development, where they are tested in humans for safety and efficacy to inform approval. Across all therapeutic areas, only about 8–12% of drugs that enter clinical trials eventually receive FDA approval.
Figure 2. The drug discovery funnel narrows toward the clinical testing stage. From: Preziosi, 2004.
In principle, we can improve productivity at any stage of this funnel: we can raise the quality of inputs through better science, relax regulatory barriers at the end, or accelerate the middle part of clinical development. Yet it’s striking how little public attention focuses on the practicalities of clinical development, despite its importance. This is the stage where theoretical promise is tested against reality: where we discover whether a biological idea can become a safe and effective therapy. It is also the most resource-intensive phase, accounting for roughly 60–70% of drug development costs and timelines.
Because clinical development is so important, it makes little sense to focus attention only on the top and bottom of the drug discovery funnel. Even as basic science and AI advance rapidly, it is unlikely that in the coming decades they will substitute for evidence gathered directly in humans. Clinical trials, with all the financial burden they bring and ethical questions they raise, will remain the critical bottleneck in translating biological insight into real therapies.
Likewise, loosening approval standards is insufficient. In fact, the FDA has already become more permissive in its approval over the past 30 years, increasingly accepting surrogate endpoints in place of demonstrated clinical benefit. But we don’t just want to approve more drugs — we want to identify and deliver drugs that actually work. Even if approval requirements were relaxed, the underlying need would remain: we still have to learn, through testing in humans, which treatments are effective and which are not.
The case for optimizing clinical trials becomes even clearer when we consider the underlying economics of drug discovery. Not only do most drug candidates fail to reach approval, but even among those that do, only about half ever generate meaningful revenue. And within this small subset of commercial successes, only a handful — the so-called blockbuster drugs, such as GLP-1 agonists or the anti-tumor necrosis factors (TNFs) —are truly transformative.
Drug discovery outcomes follow a heavy-tailed distribution: most efforts, even those deemed successful, produce modest results, while a few outliers account for a disproportionate share of both clinical and economic value. Given that success is rare, hard to predict, and potentially enormous, we need to maximize our shots on goal to increase our chances of success. More concretely, that means scaling the number of molecules tested in humans. Expanding the capacity for in-human testing broadens the exploration, increasing the likelihood of uncovering rare, high-impact breakthroughs that drive the most meaningful form of biomedical progress.
While we don’t really know how quickly better science will sharpen our therapeutic hypotheses, we have evidence that clinical development can be faster and less expensive. In Australia, Phase I trials are completed 40–50% faster and at significantly lower cost than in the US, despite comparable ethical and safety standards. In China, new drugs often reach first-in-human testing within ~18 months, versus multi-year timelines in the US. This clinical nimbleness has been often cited as a key factor behind China’s rapid progress in biotech, which is now threatening the long-standing supremacy of the US in the industry.
For the US, the ideal strategy lies in combining world-class science with highly agile clinical development. Yet clinical development has long been overshadowed by basic research, largely because it is operational, less glamorous, and thus, poorly suited for study within academic frameworks. This persistent asymmetry in attention must be addressed.
More science won’t be enough
Techno-optimists think better basic science can reverse Eroom’s Law in two ways. It can boost predictive validity so we know much earlier which drugs are likely to work, and it can unlock new kinds of therapies that finally reach targets we couldn’t hit before. In this view, a more accurate understanding of biology, from comprehensive cell atlases to organoids and AI-driven disease models, will allow researchers to navigate drug discovery with far greater precision. At the same time, emerging modalities (or tools) such as gene editing, RNA therapeutics, and targeted protein degraders will widen the spectrum of actionable biology. Together, these advances point toward a future in which each drug discovery effort has a substantially higher chance of success.
We agree that improving our therapeutic tools and our understanding of biology will be important for reversing Eroom’s Law. But we remain skeptical that scientific progress alone will make extensive human trials unnecessary in the coming decades.
The promise and perils of maps
In his novel The Glass Bead Game, Herman Hesse describes Castalia, a scholarly world devoted to the refinement of symbolic systems. Its practitioners become so absorbed in the elegance of their constructions that they forget the symbols were ever meant to point to anything outside themselves. The map becomes the world.
Something similar can happen in the life sciences. Biology is messy, dynamic, and nonlinear, yet scientists naturally gravitate toward models that make it seem orderly and tractable. A clean mechanistic pathway or target-based narrative is deeply appealing: it suggests that cause and effect are simple, knowable, and controllable. As our analytical tools improve, these explanations become more elaborate.
Through multiple waves of technology, from computer-aided drug design, via high-throughput screening, recombinant proteins, and genomics, techno-optimists have overestimated the innovation yield of the hot new thing. Again and again, scientists have placed too much confidencein the power of “biological insights,” or pre-clinical mechanistic foresight. Attention naturally concentrates on the few drugs that succeed, so it is easy to construct post hoc narratives of deliberate design.
Moreover, the biotech ecosystem rewards storytelling. From venture capital pitch decks to internal R&D reviews, a compelling mechanistic narrative makes a program easier to fund and justify.
Yet the empirical record shows that mechanistic foresight provides, at best, rough guidance. Drug discovery is better seen as an iterative design-make-test loop, in which real-world human data repeatedly guide the next cycle of design. Progress may depend less on hitting the best therapeutic hypothesis from the start, and more on generating a broad range of plausible attempts and winnowing them quickly based on clinical feedback. What works survives; what does not is modified or abandoned.
In previous work, we described this dynamic as clinical selection: a process in which the clinic, rather than preclinical mechanistic theory, supplies the decisive information about which interventions genuinely benefit patients. We contrasted this with the familiar “intelligent design” narrative, which imagines a linear march from target identification to rational design to cure.
Many of the most successful drugs did not emerge from deep mechanistic foresight, but from iterative, empirical exploration. The clinic functioned as an evolutionary engine. Anti-TNF drugs failed in their original indication before becoming foundational in autoimmune disease; statins survived only because physicians noticed striking patient responses after the field had largely moved on; and drugs like Avastin and Gleevec accumulated unexpected indications as human studies reshaped both their use and their mechanistic stories over time.
GLP-1 agonists offer a contemporary case in point. The earliest drugs in this class, such as exenatide, were developed for diabetes and aimed primarily at improving glycemic control. Later agents like liraglutide offered better pharmacological characteristics, making weight-loss applications more feasible. Even so, many experts thought that meaningful weight reduction was unattainable, because it required higher doses that caused unacceptable nausea. That side effect was overcome through clinical experimentation: gradual dose escalation markedly improved tolerability, enabling liraglutide’s approval for obesity in 2014.
Once a strong clinical signal existed, investment shifted back to refining the molecules themselves. Through extensive screening and chemical optimization of stability, potency, and half-life, Novo Nordisk developed semaglutide, a more durable agent suitable for weekly dosing. Clinical experimentation in patients without diabetes then delivered another surprise: patients lost far more weightthan most experts predicted. At higher doses, semaglutide showed ~12.4% weight loss baseline body weight vs. placebo, a result that had previously been seen as out of reach for pharmaceutical interventions.
On the back of these results, semaglutide became one of the most commercially and clinically successful medicines of the modern era. Ongoing trials continue to reveal additional, unforeseen benefits of GLP-1 agonism, including reductions in cardiovascular events that appear independent of weight loss, as well as improvements inliver disease.
Only in the last couple of years have researchers started tostitch together a more complete mechanistic picture of GLP-1–driven weight loss, integrating evidence from animal studies, neuroimaging, gut–brain signalling, adipose-tissue biology, liver metabolism, and long-duration receptor pharmacology. Crucially, this understanding emerged after, not before, the clinical breakthroughs. And the mechanistic model remains incomplete, while the clinical outcomes are unambiguous, a clear case where human trials revealed the therapeutic potential before mechanistic biology could explain it.
The incredible arc of GLP-1 agonists also highlights how powerful the profit feedback loop can be when it is aligned with clinical success: strong therapeutic effect attracts investment, investment fuels optimization, and optimization yields even greater patient benefit. By contrast, when investment is decoupled from demonstrated clinical benefit, capital can flow into drugs that fail to meaningfully help patients.
A broad historical lens reinforces the point. As described in The Rise and Fall of Modern Medicine, the mid-1940s to 1970s were characterized by a tight feedback loop between new chemistry and human experimentation. Entirely new molecular classes, antibiotics, corticosteroids, and antihypertensives, were tested rapidly in patients. Clinical selection proved remarkably effective, despite our limited biological understanding. This period remains the most productive era in pharmaceutical innovation, earning the label “the Golden Era of drug discovery”.
So far, our arguments have relied in large part on extrapolating from historical precedent. But what if this time really is different? AI is often cited as the reason it might be.
We share the optimism that AI will improve efficiency across many stages of drug development, from target discovery to trial design. However, it is unlikely (at least in the coming decades) to eliminate the need for empirical testing in humans. Indeed, some AI company CEOs have identified the inefficiency of clinical trialsas a key regulatory barrier to allowing the benefits of AI to spill over into biomedicine.
Whether AI models can replace clinical testing ultimately depends on the data they are trained on. AI has achieved remarkable success in biology when applied to problems that are well-constrained and richly parameterized; where large, high-quality datasets exist, and the mapping between inputs and outputs is tight. AlphaFold’s success in protein structure prediction is a paradigmatic example, as it addressed a closed system with extensive labeled data and well-defined ground truth.
By contrast, the central challenge in drug development, the translation of molecular intervention into complex, organism-level therapeutic effects, remains underdetermined. For most disease areas, the relevant data landscape is sparse, heterogeneous, and observational. Perhaps the richest forms of relevant data are large-scale multi-omic datasets. These integrate multiple layers of biological information, such as genomic, transcriptomic, proteomic, metabolomic, and epigenomic profiles, to map how molecular systems interact across scales. Yet, despite their richness, they are taken at single timepoints and fail to capture the dynamic feedback loops, nonlinearities, and stochasticity that characterize living systems.
While such efforts are important for the long term, they are unlikely to replace empirical testing, unless dramatically novel modes of data generation that reflect human physiology are developed. This is where increasing the number of clinical trials could actually complement the predictive power of AI models. More experimental medicine in humans is not a substitute for AI, but its informational and economic complement: it would generate the data that would make AI more predictive over time.
Widen the funnel, instead of approving more low-quality drugs
Contrary to the libertarian view, loosening the standards for approval won’t fix the problem. Their logic seems appealing: if the regulatory bar is lowered, more therapies will reach patients sooner. And in some cases, we agree that approval standards should indeed be relaxed. But by focusing on the end of the pipeline rather than the process of experimentation itself, such reforms risk admitting more weakly effective or ineffective drugs, while doing little to expand the discovery of genuinely transformative ones.
Lowering the approval burden would, by definition, reduce clinical development costs: if fewer trials are required, sponsors face fewer expenses. Economist Alex Tabarrok has gone further, arguing that the FDA should only evaluate safety, leaving questions of efficacy to be resolved through post-approval clinical use. In such a system, the low initial hurdle would naturally encourage more drugs to be brought forward due to the lower up-front investment needed.
There is historical precedent for new use cases for drugs emerging from real-world use as opposed to randomized controlled trials. But there are many diseases, especially chronic ones with slow and noisy progression, where we do not envision how high-quality data on therapeutic effect can be collected without rigorous trials.
Moreover, even if the FDA were to approve all drugs that are merely safe, questions around insurance coverage and reimbursement would remain. In such a scenario, private entities may emerge to evaluate efficacy. But if clinical trials remained costly and time-consuming, easing regulatory requirements at the approval stage alone would do little to reduce the burden of generating credible evidence.
Over the past three decades, the FDA has become more permissive in what it accepts as evidence for drug approval. Between 1995 and 2017, pivotal trials grew less methodologically rigorous, with declines in randomization, blinding, the use of active comparators, and the measurement of real clinical outcomes. Most notably, trials have increasingly relied on surrogate endpoints that predict patient benefit rather than demonstrate it directly. This shift enables faster and cheaper studies, but it also raises concerns about whether approved drugs provide meaningful benefits for patients in the real world.
For a dramatic case in point, look at the approval of aducanumab for Alzheimer’s disease in 2021 through the accelerated approval pathway, based on beta-amyloid reduction as an endpoint. Despite two Phase III trials failing to show cognitive benefit, broad dissent within the FDA regarding its approval, and existing meta-analyses showing a poor correlation between beta-amyloid reduction and meaningful clinical benefit, the FDA granted accelerated approval. The result was a$56,000-per-year therapy with uncertain benefit, triggering significant backlash and eroding public trust. Congressional investigations and internal FDA reviews later concluded that the decision lacked scientific justification and damaged the agency’s credibility.
Support for easing approval standards is not limited to libertarian economists. Patient advocacy groups, especially in rare diseases, have increasingly pushed for similar reforms. These groups have not pushed as hard on inefficiencies in the clinical process in part because the process is opaque, while approval decisions are public, dramatic, and easy to mobilize around. But in clinical development, everything from details around trial design to regulatory correspondence remains largely hidden behind company walls.
Conclusion
The central paradox of modern biomedicine is that our ability to design interventions has advanced faster than our ability to test them. We are living through an era of unprecedented biological insight and tools, yet the process that determines which ideas translate into real therapies has become slower, costlier, and narrower.
Restoring exploratory capacity requires a rebalancing of the system: recognizing that clinical trials are not a bureaucratic middle step between discovery and approval, but the core engine of therapeutic learning. The Golden Era of Medicine illustrates what this looks like when it works.
The imbalance between the investment and return on clinical trials implies substantial low-hanging fruit: inefficiencies that persist not out of necessity, but neglect. But global differences in trial speed and cost, the demonstrated advantages of adaptive and platform trials, and emerging efforts to enhance regulatory transparency collectively show that safer, faster, and more informative clinical learning is well within reach.
I would add one observation about the idea of the FDA only evaluating "safety." It's not clear what that even means. Sure, we should screen out cases like TGN1412 that send everyone to the hospital with septic shock. But there are many drugs (cancer drugs in particular) that typically aren't "safe" per se. In many cases, they have side effects that are not safe at all. The only reason to ever take such drugs is because the potential benefits outweigh the serious risks, which requires knowing something about efficacy.
It’s surprising that more people aren’t talking about public trust erosion wrt opening the “right to try/pass safety” floodgates. If we think it’s bad now (especially with vaccines that work) wait until the majority of approved assets do absolutely nothing. Anyway, off my soapbox.
It is kind of funny hearing all the techno-optimists act like one more preclinical candidate is all we really need. Just one more bro. Sure there are thousands of shelved assets that have no path through trialing because it’s so expensive, time consuming, and burdensome, but all we really need is one more. As you point out, better candidates will certainly help but they aren’t the only thing!
China is very fast at getting clinical data and part of that is because of their large population and access to a ton of treatment-naive patients. If the US would play nice with allies and take the lead on trial/data standardization we could also have access to similar patient populations and so could developers in other countries.




Great piece!
I would add one observation about the idea of the FDA only evaluating "safety." It's not clear what that even means. Sure, we should screen out cases like TGN1412 that send everyone to the hospital with septic shock. But there are many drugs (cancer drugs in particular) that typically aren't "safe" per se. In many cases, they have side effects that are not safe at all. The only reason to ever take such drugs is because the potential benefits outweigh the serious risks, which requires knowing something about efficacy.
Fantastic work.
It’s surprising that more people aren’t talking about public trust erosion wrt opening the “right to try/pass safety” floodgates. If we think it’s bad now (especially with vaccines that work) wait until the majority of approved assets do absolutely nothing. Anyway, off my soapbox.
It is kind of funny hearing all the techno-optimists act like one more preclinical candidate is all we really need. Just one more bro. Sure there are thousands of shelved assets that have no path through trialing because it’s so expensive, time consuming, and burdensome, but all we really need is one more. As you point out, better candidates will certainly help but they aren’t the only thing!
China is very fast at getting clinical data and part of that is because of their large population and access to a ton of treatment-naive patients. If the US would play nice with allies and take the lead on trial/data standardization we could also have access to similar patient populations and so could developers in other countries.